Quick description¶
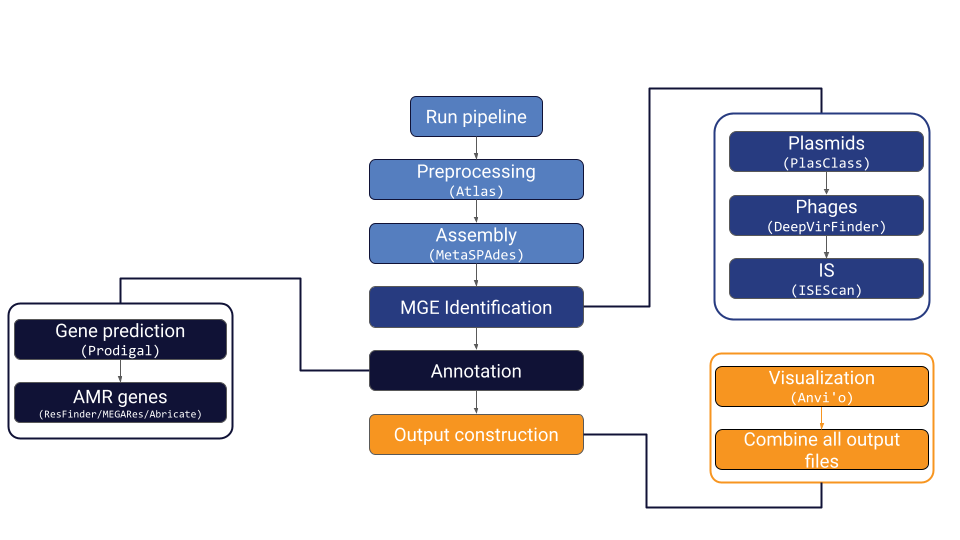
MetaMobilePicker is a Snakemake pipeline designed to identify mobile genetic elements (MGEs), specifically plasmids, insertion sequences (IS) and phages, and antimicrobial resistance (AMR) genes in metagenomics samples. It runs preprocessing steps, metagenomics assembly, several MGE identification tools, two AMR database lookups and combines the output together into one YAML file. Additionally, it creates two essential Anvi’o files for visualization purposes.
Quick installation guide¶
The quickest way to install MetaMobilePicker is to create a conda environment and install using mamba:
> conda create --name metamobilepicker python=3.10
> conda activate metamobilepicker
> conda install mamba
> mamba install -c bioconda metamobilepicker
> conda install -c bioconda biopython
Note: At this point in time, biopython needs to be installed manually. To run the pipeline you will need Singularity installed on your system with version number 3.7 or higher. If Singularity is not installed, it can be installed in one of the ways described here.
After installation, the fastest way to test the installation is to use the included test data. This dataset consists of 10.000 reads and should run relatively fast. To test the pipeline, run the following commands
> metamobilepicker run --dryrun --test
Running the pipeline¶
Within the repository the main script (metamobilepicker.py) can be found in the MetaMobilePicker directory. This is the file that runs when using the metamobilepicker command. It is not fit to run as standalone (as of version 0.6)!
Metamobilepicker.py¶
The start script has two callable submodules.
config
run
Config¶
The config module generates a config file (see “config file” for more info). This step needs to be performed before running the pipeline.
Usage: metamobilepicker config [OPTIONS]
Options:
-s, --samples TEXT Supply a custom text file with sample names and read
locations [default: samples.txt]
-a, --assembly TEXT Supply custom assembly for all samples in samples.txt
-o, --output TEXT Name of the config file
-t, --threads INTEGER Maximum number of threads to use [default: 16]
-h, --host TEXT Fasta file with host sequences
-m, --memory INTEGER Set the maximum available memory (make sure to have
enough for your assembly!) [default: 8]
-d, --datadir TEXT Set the path to your data [default: current directory]
-O, --outdir TEXT Set the path to your output directory [default: current directory]
--help Show this message and exit
To make use of this feature, first create a samples file. This is a comma-separated file (CSV) containing the names, path to forward reads and path to reverse reads per sample you want to run. Example of a samples file (named here: samples.csv. By default, metamobilepicker expects a file with the name samples.txt)
testsample,../data/raw/ERR2241639_1_test.fastq.gz,../data/raw/ERR2241639_2_test.fastq.gz
Running
metamobilepicker.py config --samples samples.csv
will create a config file from the samples files that can be used to run the pipeline.
Run¶
The run module runs the pipeline.
Usage: metamobilepicker run [OPTIONS]
Runs the MetaMobilePicker pipeline.
Options:
-n, --dryrun Test the script without running the pipeline
-a, --assembly TEXT Run MetaMobilePicker with a preexisting assembly
file
-A, --assemblyfile TEXT Run MetaMobilePicker with a set of preexisting
assemblies
-s, --snakefile TEXT Change the name of the Snakefile to be used
[default: Snakefile]
-p, --profile TEXT Use Snakemake profile
-c, --config TEXT Specify config file [default: config/config.yaml]
-u, --unlock Unlock directory after failed Snakemake attempt
-t, --test Run MetaMobilePicker with a small test set
-C, --cores INTEGER Specify the number of available cores [default:
16]
--help Show this message and exit.
By default, the pipeline will take the basic config file in the config directory. However we recommend to specify a config file generated with the config submodule and specified below. To use a premade assembly (or set of assemblies for multiple samples), the -a and -A parameters can be used. To have multiple assemblies as input, supply a text file with all paths to the assemblies on new lines. The pipeline will combine them with the samples in the order they are specified in the assembly file.
If something goes wrong while running the pipeline before an error can be displayed, like being disconnected from a server, Snakemake will lock the directory. To unlock the directory to try again, use the –unlock or -u flag.
The Config file¶
MetaMobilePicker uses the Snakemake framework to run. Config files are therefore in the YAML format. The MetaMobilePicker config files include the samples on which to run the pipeline, some information on the host contamination database and information on how many cores can be used in the process. An example can be found in the test directory
datadir: /current/directory/
host: /current/directory/human_genome.fna
max_mem: 8
outdir: /current/directory/
samples:
test:
fwd: data/sample_R1.fastq.gz
rev: data/sample_R2.fastq.gz
threads:
big: 8
huge: 16
medium: 8
small: 4
All samples are placed within the samples section with their forward (fwd) and reverse (rev) read paths. In the threads section, there are four ‘sizes’ of jobs with the number of CPUs that correspond to them. If the system does not have this amount of CPUs, Snakemake will use whatever is available. The host section contains the path to a FASTA file with host sequences. The max_mem section contains the maximum amount of memory that can be used (in Gigabytes) so that tools like Atlas know how much they can ask for. This config file can be manually created or created using the config module of the pipeline.
Output¶
The output of the pipeline is stored in a per-sample folder in the directory specified in the configfile as ‘outdir’.
In the sample results folder, there are several subdirectories. First, there is the ATLAS preprocessing folder. This folder contains the preprocessing output files, most importantly the quality-checked (QC) reads. The next subdirectory is the MetaSPAdES directory which contains all output files from the metagenomics assembly, including the contigs file with sequences larger than 1000 bp. In the “MGEs” folder, the result for all the MGE identification steps can be found. Also, the final YAML file of the pipeline (called $sample_metamobilepicker.out) along with the FASTA file containing al putative MGEs can be found in this directory. The annotation folder contains the AMR gene annotation results and the gene prediction results. The gene predictions are unused at the moment but could be used for functional annotation. The ANVIO directory contains a contigs and profile database that can be used for further analyses using Anvi’o. In the mapping directory, BAM files containing the alignment of the reads to the assembly are located.
Final output file¶
The final output file of MetaMobilePicker is a custom output file that can be parsed like a YAML file. Example:
contig:
contig_id: NODE_2_length_12828_cov_50.403664
length: 12828
number_IS: 1
number_annotations: 2
plasmid:
ID: plasmid
score: 0.966940194363968
Annotation_1:
name: Drugs:Glycopeptides:VanA-type_regulator:VANRA
start: 235
stop: 720
source: megares
gene: VANRA
accession: MEG_7452
Annotation_2:
name: Drugs:Glycopeptides:VanA-type_regulator:VANRA
start: 873
stop: 1783
source: megares
gene: VANRA
accession: MEG_7458
IS_1:
ID: IS6_292
class: IS6
start: 2107
stop: 2921
This format allows for hierarchical annotation of contigs with optional fields like ISs and AMR genes.
Additionally, a FASTA file is generated containing only the contigs predicted as plasmid or phage, or that have an IS annotated with at least 200bp on either side of the IS. The removal of IS with flanking regions shorter than 200bp is to make sure there is information on the contig other than the IS that can be used for further analysis. The details of the annotations and classifications can be found in the FASTA header.
Tutorial¶
Check if the installation of MetaMobilePicker was successful using this tutorial.
Testing the Pipeline¶
Technical test¶
After installation, the fastest way to test the installation is to use the included test data. This dataset consists of 5.000 reads and should run relatively fast. To test the pipeline, run the following commands
> metamobilepicker run --test --dryrun
If this doesn’t give any errors, run the pipeline with the following command
> metamobilepicker run --test
If this is the first run of the pipeline, it will create the appropriate conda environments and download the used containers, which can take a while.
Testing a run from scratch¶
To make sure everything is working as intended, you can create a new run using the same test data.
Config files¶
MetaMobilePicker is a Snakemake pipeline that works with YAML config files. The easiest way is to let MetaMobilePicker generate its own config file. Before we can do this, we need to generate the samples file. This comma separated file contains your sample names and the paths to the paired end reads. Before making the config file, copy the test reads to a location where you can easily locate them. In your prefered directory run the following commands
> mkdir mmp_data
> cp {PATH TO REPOSITORY}/MetaMobilePicker/test/test_reads_R1.fastq mmp_data
> cp {PATH TO REPOSITORY}/MetaMobilePicker/test/test_reads_R2.fastq mmp_data
> mkdir mmp_test_output # Our output files will go here
Now we can create our samples.txt file to look like this
testsample,mmp_data/test_reads_R1.fastq,mmp_data/test_reads_R2.fastq
Save this file as samples.txt for now. Next, we generate the config file
> metamobilepicker config --samples samples.txt --output test_config.yaml --outdir mmp_test_output
This should give you a file in the current directory called test_config.yaml that contains all the information we need to run MetaMobilePicker.
Running the pipeline using our config file¶
Next, to test the installation of the pipeline, run the following command
> metamobilepicker run -c test_config.yaml --dryrun
If this doesn’t give errors, go ahead and run
Troubleshooting¶
MetaMobilePicker depends on the (conda) installation of several tools. This can lead to unexpected errors when trying to install all environments on a different system. Here we show some issues we found during testing.
Installing the Anvi’o environment
From the Anvi’o documentation: While setting up your environment to track the development branch, especially on Ubuntu systems (first observed on Ubuntu 20.04 LTS), you may run into issues related to package conflicts that produce error messages like this one:
Encountered problems while solving:
- nothing provides r 3.2.2* needed by r-magrittr-1.5-r3.2.2_0
- nothing provides icu 54.* needed by r-base-3.3.1-1
- package sqlite-3.32.3-h4cf870e_1 requires readline >=8.0,<9.0a0, but none of the providers can be installed
- package samtools-1.9-h8ee4bcc_1 requires ncurses >=6.1,<6.2.0a0, but none of the providers can be installed
These problems can be solved by explicitly setting conda with flexible channel priority setting. Run these commands to change the channel priority setting:
conda config --describe channel_priority
conda config --set channel_priority flexible
And re-run the commands to install conda packages. You can set the priority back to ‘strict’ at any time.
Singularity issues The installation of Singularity can be the source of some bugs. When you are not the administrator of the system you’re trying to instal MetaMobilePicker on, it is important to first check if the administrator has a system-wide version of Singularity installed. This can be checked using the following command
If this command gives an error, Singularity is not installed. Please try to install it from the singularity website or from conda.
Please note that the minimal required Singularity version for MetaMobilePicker is v3.7.
Contributions¶
We welcome contributions in the form of GitLab issues or pull requests, or additions to this documentation.