General overview¶
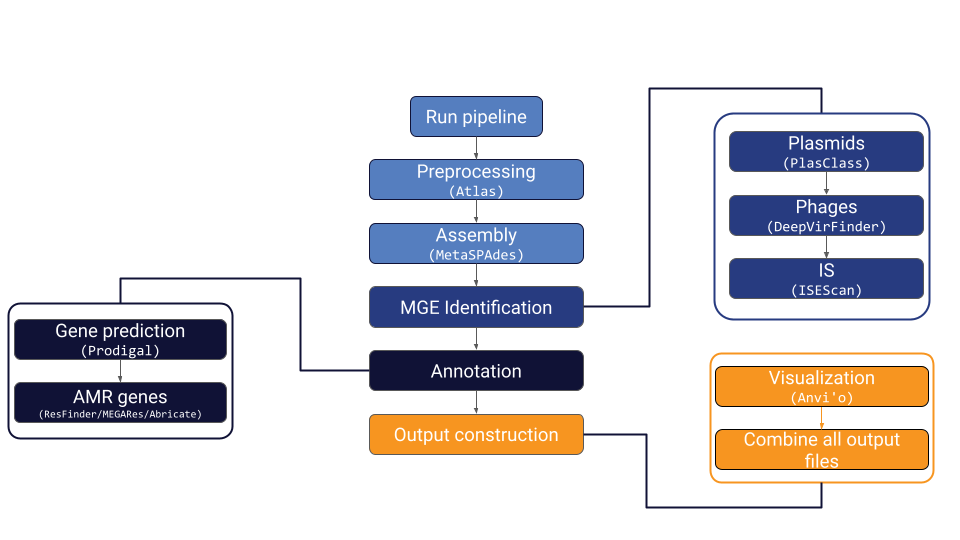
MetaMobilePicker identifies mobile genetic elements (MGEs) and annotates antimicrobial resistance genes (ARGs) from metagenomic short reads. MetaMobilePicker can be roughly divided into three modules: Preprocessing, MGE Identification, Annotation and Output Construction.
Preprocessing¶
During the preprocessing steps, the metagenomic short reads are subjected to quality control (QC) and metagenomics assembly. QC is done using the QC Module of Metagenome Atlas (Kieser et al. [2020]). This tool performs deduplication, quality trimming and host contamination removal. Metagenomics assembly is done using MetaSPAdes (Nurk et al. [2017]). Contigs larger than 1000 bp are being used for the following modules.
MGE Identification¶
During the MGE identification step, plasmids, phages and insertion sequences (IS) are identified using PlasClass (Pellow et al. [2020]), DeepVirFinder (Ren et al. [2020]) and ISEScan (Xie and Tang [2017]) respectively. For plasmids, contigs with a plasmid score larger than 0.8 are retained as plasmids. For phages, contigs with a score larger than 0.95 are retained as phage. For IS, default annotation parameters are used.
Annotation¶
During the Annotation steps, Abricate (https://github.com/tseemann/ABRicate) is used to annotate ARGs using the ResFinder (Florensa et al. [2022]) and MEGARES (Doster et al. [2020]) databases. Additionally, as a precursor for further functional annotation, Prodigal (Hyatt et al. [2010]) is used for gene prediction.
Output Construction¶
During the output construction steps, custom scripts are used to create a YAML file containing the combined output of the previous modules. Additionally a fasta file containing only the predicted MGEs is created with the annotation information in the fasta headers. Contigs containing IS are filtered where contigs containing less than 200bp outside the IS are discarded. Using BWA (Li and Durbin [2009]), the quality controled reads are aligned to the fasta containing the MGEs. The resulting bam file is used to create a profile and contig db using Anvi’o (Eren et al. [2021]) for further visualization and annotation. By default, Anvi’o uses hierarchical clustering on the contigs to make a visualizable dendogram. This should usually not create issues due to the limited number of MGEs in a sample. However, if it does cause issues, please create a GitLab issue.
Note on processing power¶
MetaMobilePicker is a relatively heavy pipeline, since it includes computationally demanding steps such as metagenomics assembly. We therefore recommend to run the pipeline on an HPC or a large server that is otherwise capable of running a metagenomics assembly, with a minimum of 64GB of RAM. MetaMobilePicker is as parallel as possible, using Snakemake (Mölder et al. [2021]) as a job distribution backbone, and facilitates the use of Snakemake profiles for HPC job submission, but theoretically the tool could run on a low number of threads.
Benchmarking data¶
To get an idea of the time and resources required to run MetaMobilePicker efficiently, we ran the tool on 20 metagenomics samples with 40M reads. Below are the average time and resource usages. Using higher numbers of CPUs can decrease the amount of time it takes to run a sample, but can lead to an increase of memory usage.
Tool |
Provided cores |
Memory usage |
CPU time (h:m) |
|---|---|---|---|
ATLAS QC |
16 |
50G |
6:19 |
MetaSPAdes |
24 |
123G |
9:46 |
PlasClass |
16 |
19.7G |
0:6 |
DeepVirFinder |
16 |
30G |
5:19 |
ISEScan |
16 |
500M |
12:44 |
References¶
Enrique Doster, Steven M. Lakin, Christopher J. Dean, Cory Wolfe, Jared G. Young, Christina Boucher, Keith E. Belk, Noelle R. Noyes, and Paul S. Morley. MEGARes 2.0: a database for classification of antimicrobial drug, biocide and metal resistance determinants in metagenomic sequence data. Nucleic Acids Research, 48(D1):D561–D569, January 2020. doi:10.1093/nar/gkz1010.
A. Murat Eren, Evan Kiefl, Alon Shaiber, Iva Veseli, Samuel E. Miller, Matthew S. Schechter, Isaac Fink, Jessica N. Pan, Mahmoud Yousef, Emily C. Fogarty, Florian Trigodet, Andrea R. Watson, Özcan C. Esen, Ryan M. Moore, Quentin Clayssen, Michael D. Lee, Veronika Kivenson, Elaina D. Graham, Bryan D. Merrill, Antti Karkman, Daniel Blankenberg, John M. Eppley, Andreas Sjödin, Jarrod J. Scott, Xabier Vázquez-Campos, Luke J. McKay, Elizabeth A. McDaniel, Sarah L. R. Stevens, Rika E. Anderson, Jessika Fuessel, Antonio Fernandez-Guerra, Lois Maignien, Tom O. Delmont, and Amy D. Willis. Community-led, integrated, reproducible multi-omics with anvi’o. Nature Microbiology, 6(1):3–6, January 2021. Number: 1 Publisher: Nature Publishing Group. URL: https://www.nature.com/articles/s41564-020-00834-3 (visited on 2022-11-03), doi:10.1038/s41564-020-00834-3.
Alfred Ferrer Florensa, Rolf Sommer Kaas, Philip Thomas Lanken Conradsen Clausen, Derya Aytan-Aktug, and Frank M. Aarestrup. ResFinder – an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microbial Genomics, 8(1):000748, January 2022. URL: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8914360/ (visited on 2022-11-15), doi:10.1099/mgen.0.000748.
Hadrien Gourlé, Oskar Karlsson-Lindsjö, Juliette Hayer, and Erik Bongcam-Rudloff. Simulating Illumina metagenomic data with InSilicoSeq. Bioinformatics, 35(3):521–522, February 2019. URL: https://doi.org/10.1093/bioinformatics/bty630 (visited on 2022-05-03), doi:10.1093/bioinformatics/bty630.
Doug Hyatt, Gwo-Liang Chen, Philip F. LoCascio, Miriam L. Land, Frank W. Larimer, and Loren J. Hauser. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11(1):119, March 2010. URL: https://doi.org/10.1186/1471-2105-11-119 (visited on 2022-11-03), doi:10.1186/1471-2105-11-119.
Silas Kieser, Joseph Brown, Evgeny M. Zdobnov, Mirko Trajkovski, and Lee Ann McCue. ATLAS: a Snakemake workflow for assembly, annotation, and genomic binning of metagenome sequence data. BMC Bioinformatics, 21(1):257, June 2020. URL: https://doi.org/10.1186/s12859-020-03585-4 (visited on 2022-05-03), doi:10.1186/s12859-020-03585-4.
Heng Li and Richard Durbin. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England), 25(14):1754–1760, July 2009. doi:10.1093/bioinformatics/btp324.
Felix Mölder, Kim Philipp Jablonski, Brice Letcher, Michael B. Hall, Christopher H. Tomkins-Tinch, Vanessa Sochat, Jan Forster, Soohyun Lee, Sven O. Twardziok, Alexander Kanitz, Andreas Wilm, Manuel Holtgrewe, Sven Rahmann, Sven Nahnsen, and Johannes Köster. Sustainable data analysis with Snakemake. F1000Research, 10:33, 2021. doi:10.12688/f1000research.29032.2.
Sergey Nurk, Dmitry Meleshko, Anton Korobeynikov, and Pavel A. Pevzner. metaSPAdes: a new versatile metagenomic assembler. Genome Research, 27(5):824–834, May 2017. URL: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5411777/ (visited on 2022-05-03), doi:10.1101/gr.213959.116.
David Pellow, Itzik Mizrahi, and Ron Shamir. PlasClass improves plasmid sequence classification. PLOS Computational Biology, 16(4):e1007781, April 2020. Publisher: Public Library of Science. URL: https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007781 (visited on 2022-05-03), doi:10.1371/journal.pcbi.1007781.
Jie Ren, Kai Song, Chao Deng, Nathan A. Ahlgren, Jed A. Fuhrman, Yi Li, Xiaohui Xie, Ryan Poplin, and Fengzhu Sun. Identifying viruses from metagenomic data using deep learning. Quantitative Biology, 8(1):64–77, March 2020. URL: https://doi.org/10.1007/s40484-019-0187-4 (visited on 2022-05-03), doi:10.1007/s40484-019-0187-4.
Alice R. Wattam, David Abraham, Oral Dalay, Terry L. Disz, Timothy Driscoll, Joseph L. Gabbard, Joseph J. Gillespie, Roger Gough, Deborah Hix, Ronald Kenyon, Dustin Machi, Chunhong Mao, Eric K. Nordberg, Robert Olson, Ross Overbeek, Gordon D. Pusch, Maulik Shukla, Julie Schulman, Rick L. Stevens, Daniel E. Sullivan, Veronika Vonstein, Andrew Warren, Rebecca Will, Meredith J.C. Wilson, Hyun Seung Yoo, Chengdong Zhang, Yan Zhang, and Bruno W. Sobral. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Research, 42(Database issue):D581–D591, January 2014. URL: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3965095/ (visited on 2022-11-14), doi:10.1093/nar/gkt1099.
Zhiqun Xie and Haixu Tang. ISEScan: automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics (Oxford, England), 33(21):3340–3347, November 2017. doi:10.1093/bioinformatics/btx433.